Миопатия дюшенна: причины, симптомы, лечение
Прогрессирующая мышечная дистрофия Дюшенна — наследуемая сцеплено с Х-хромосомой патология мышечной системы, проявляющаяся в первые 3-5 лет жизни и характеризующаяся быстро распространяющейся и усугубляющейся мышечной слабостью. Первоначально поражаются мышцы тазового пояса и бедер, затем — плеч и спины, постепенно наступает обездвиженность. Миодистрофия сопровождается скелетными деформациями и поражением сердца. Диагностика дистрофии Дюшенна включает неврологическое и кардиологическое обследование, определение уровня КФК, электромиографию, консультацию генетика, ДНК-анализ, биопсию мышц. Лечение симптоматическое. В связи со слабостью дыхательной мускулатуры на заключительном этапе заболевания требуется ИВЛ.
Содержание
- Прогрессирующая мышечная дистрофия Дюшенна
- Причины мышечной дистрофии Дюшенна
- Симптомы мышечной дистрофии Дюшенна
- Диагностика мышечной дистрофии Дюшенна
- Лечение мышечной дистрофии Дюшенна
- 6 распространенных форм миопатии
- Что представляет собой миопатия
- Чем грозит полиневропатия
- Классификация
- Узнаем, почему немеют пальцы левой руки
- Диагностика
- Последствия и осложнения
- Миопатия Дюшена
- Краткая характеристика миопатии Дюшена
- Симптомы заболевания
- Диагностика
- Лечение миопатии Дюшена
- Профилактика
- Миопатия Дюшенна: симптомы и лечение
- Симптоматика
- Диагностика
- Возможные осложнения
- Профилактика
- Особенности развития и лечения миопатии Дюшена
- Почему возникает патология?
- Как проявляется заболевание?
- В чем опасность патологии?
- Каковы способы диагностики?
- Как лечится патология?
Прогрессирующая мышечная дистрофия Дюшенна
Причины мышечной дистрофии Дюшенна
Развитие мышечной дистрофии Дюшенна связано с наличием мутации в 21-ом локусе короткого плеча Х-хромосомы в гене, кодирующем белок дистрофин. Около 70% случаев болезни вызваны дефектным геном дистрофина, полученным от матери — носительницы патологической мутации. Остальные 30% связаны с появлением свежих мутаций в яйцеклетках матери. В отличие от миодистрофии Беккера, при дистрофии Дюшенна генетические аберрации приводят к сдвигу рамки считывания ДНК и полному прекращению синтеза дистрофина, что и обуславливает более тяжелое течение патологии.
В норме входящий в сарколемму миоцитов дистрофин обеспечивает ее целостность и устойчивость к растяжению, возникающему при сократительной активности мышечных волокон. Отсутствие дистрофина влечет за собой нарушение целостности сарколеммы, разрушение миоцитов и их замещение жировой и соединительной тканью. Клинически этот процесс выражается прогрессирующим снижением способности мышц к сокращению, утратой мышечной силы и тонуса, атрофией мышц.
Симптомы мышечной дистрофии Дюшенна
Дебют миодистрофии Дюшенна приходится на период от 1 до 5 лет. Как правило, уже на 1-ом году жизни заметно некоторое отставание моторного развития ребенка. Отмечается задержка сроков начала сидения, самостоятельного вставания и ходьбы. Когда ребенок начинает ходить, он отличается неуклюжестью и большей, по сравнению со сверстниками, неустойчивостью; часто спотыкается.
Мышечная слабость возникает на 3-4-ом годах жизни. Первоначально она выражается в патологически повышенной утомляемости при ходьбе по лестнице или на длинные расстояния. Со временем становится заметной типичная для миодистрофий «утиная» походка. Обращают на себя внимание особенности поведения ребенка — каждый раз, поднимаясь из положения сидя на корточках, он активно опирается руками о собственное тело, как бы взбираясь по нему как по лесенке (симптом Говерса).
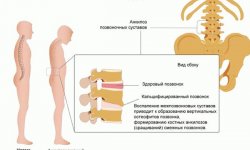
Мышечные атрофии начинаются с мышц бедер и тазового пояса. Для дистрофии Дюшенна характерно их быстрое восходящее распространение на плечевой пояс, мускулатуру спины и проксимальных отделов рук. Вследствие мышечных атрофий формируется «осиная» талия и отстоящие от спины «крыловидные» лопатки. Типичным симптомом выступает псевдогипертрофия икроножных мышц. Наблюдается выпадение сухожильных рефлексов: вначале — коленных, затем — рефлексов с трицепса и бицепса плеча. Ахилловы и карпорадиальные рефлексы могут длительное время быть сохранны. Со временем развиваются ретракции сухожилий и мышечные контрактуры.
Прогрессирующая мышечная дистрофия Дюшенна сопровождается нарушениями в костно-суставной системе. Характерны искривление позвоночника (кифоз, усиленный лордоз, сколиоз), деформации грудной клетки (килевидная или седловидная), деформации стоп. Сердечно-сосудистые расстройства обусловлены развитием кардиомиопатии и включают аритмию, лабильность артериального давления, глухость тонов сердца. У 50% больных фиксируются нейроэндокринные расстройства — адипозогенитальная дистрофия, синдром Иценко-Кушинга и др. Около 30% больных страдает олигофренией, как правило, ограничивающейся степенью дебильности. Могут отмечаться СДВГ, расстройства по типу аутизма, дислексия, нарушения краткосрочной памяти.
Уже к 7-10-летнему возрасту дистрофия Дюшенна приводит к выраженным двигательным ограничениям. К 12 годам больные, как правило, утрачивают способность ходить, а к возрасту 15 лет большинство пациентов полностью теряют возможность самостоятельных движений. Распространение дистрофического процесса на дыхательную мускулатуру приводит к прогрессирующему падению жизненной емкости легких (ЖЕЛ) и, в конечном итоге, невозможности совершать дыхательные движения.
Диагностика мышечной дистрофии Дюшенна
Установить диагноз миодистрофии Дюшенна помогает анамнез, неврологическое обследование, результаты электрофизиологического тестирования, определение креатинфосфокиназы (КФК) в биохимическом анализе крови, морфологическое и иммунохимическое исследование образцов мышечной ткани, генетическое консультирование и анализ ДНК. При этом дифференциальную диагностику следует проводить с другими миопатиями — метаболической, воспалительной, миодистрофией Беккера, мышечной дистрофией Дрейфуса, дистрофией Эрба-Рота, а также с полиневропатиями, полимиозитом, БАС.
В случаях, когда имеется клиническая картина миодистрофии, а анализ ДНК не выявил наличие мутации, показана биопсия мышц. Морфологическое исследование биоптата определяет разнокалиберность и некроз миоцитов, их замещение соединительнотканными элементами. Иммунохимический анализ говорит о полном отсутствии дистрофина в исследуемых мышечных волокнах.
Дополнительно осуществляется обследование костно-мышечной и сердечно-сосудистой систем — проводится консультация ортопеда, рентгенография позвоночника, обзорная рентгенография ОГК, консультация кардиолога, ЭКГ, эхокардиография. По показаниям рекомендуется консультация эндокринолога, пульмонолога и др. специалистов.
Лечение мышечной дистрофии Дюшенна
Терапия, применяемая в клинической практике, пока включает лишь необходимые симптоматические мероприятия. Для улучшения метаболизма мышечной ткани возможно назначение анаболических стероидов (метандиенона, нандролона деканоата), АТФ, актопротекторов (этилтиобензимидазола); для облегчения нервно-мышечной передачи — неостигмина. С целью минимизировать образование контрактур и продлить двигательную активность пациентов проводится ЛФК, массаж, физиотерапия.
Важное значение имеет контроль дыхательной функции и газового состава крови. При падении ЖЕЛ до 40% рекомендована искусственная вентиляция легких в период сна. В дальнейшем время ИВЛ растет пропорционально снижению ЖЕЛ. В начале ИВЛ может осуществляться при помощи масочного аппарата. Затем необходима трахеостомия, и ИВЛ проводится путем присоединения аппарата к трахеостомической трубке. Современные портативные аппараты ИВЛ работают на батареях и могут быть закреплены на инвалидной коляске.
Поиск эффективных способов лечения дистрофии Дюшенна — задача, над решением которой трудятся сегодня специалисты в области неврологии, биохимии, генной инженерии. Из перспективных разработок в этом направлении можно выделить лечение стволовыми клетками, активацию гена утрофина, являющегося наиболее близким аналогом дистрофина, технологию пропуска экзонов.
Прогноз и профилактика
Из всех форм миодистрофии дистрофия Дюшенна имеет наиболее неблагоприятный прогноз. Манифестация заболевания в раннем возрасте приводит к тому, что к 15 годам пациенты становятся полностью обездвижены. Летальный исход неизбежен. Зачастую больные не достигают 25-летнего возраста. Обычно смертельный исход обусловлен интеркуррентными инфекциями, застойной пневмонией, сердечной или дыхательной недостаточностью.
Профилактические мероприятия направлены на выявление женщин-носительниц аномального гена дистрофина и предупреждение рождения у них больного ребенка. В рамках профилактических мер проводятся консультации генетика для планирующих беременность супружеских пар, консультации беременных и пренатальная ДНК-диагностика.
6 распространенных форм миопатии
Различные нарушения в метаболизме, а также в построении мышечной ткани, которые приводят к ограничению двигательной активности и снижению физической силы, называют миопатиями.
К чертам данного заболевания можно отнести следующие явления:
- Нарастающая мышечная слабость.
- Слабые рефлексы сухожилий, либо их полное отсутствие.
- Мышечная атрофия.
Миопатии обуславливаются генетическими факторами, и часто встречаются у представителей одной семьи.
Что представляет собой миопатия
Миопатия характеризуется поражением ткани мышц и относится к нервно-мышечной группе заболеваний. Помимо скелетной мускулатуры, ей подвергаются и миофибриллы (отдельные волокна), при этом анимальная нервная система остается в полной сохранности.
Чем грозит полиневропатия
- Posted on Июнь 5, 2018 Июнь 1, 2018
- Маргарита Юрьевна Новикова
- Невралгия
- Нервно-мышечные заболевания
- 3 просмотра
- 6 минут на чтение
Заболевание протекает прогрессивно, приводя к дистрофии определенный участок мышечной ткани. Наиболее часто болезнь диагностируется у детей и подростков. Миопатия может быть как первичной, так и вторичной. Наиболее часто встречается форма заболевания, передаваемая по наследству. В остальных случаях мышечная дистрофия имеет характер приобретенного генеза.
Миопатию часто путают с невропатией, но разница между этими заболеваниями заключается в том, что в первом случае у больного не нарушается чувствительность конечностей. При миопатии он по-прежнему может ощущать боль, жжение, зуд, холод и жар.
Классификация
За последние десятилетия ученые-исследователи вывели следующие формы заболевания.
Миопатия Дюшенна
Данный вид заболевания характерен для пациентов 3-х летнего возраста, причем оно не распространяется на представителей женского пола. Форма болезни Дюшенна представляет серьезную опасность как для здоровья, так и для жизни больного.
Поражаются мышцы ягодиц и тазобедренного участка, что не позволяет ребенку самостоятельно передвигаться. Если способность ходить возможна, то возникают ограничения – ребенок не может подниматься по ступеням, прыгать и вставать.
Заболевание приводит к деформации костей, умственной отсталости и псевдогипертрофированию мышечной ткани икр. Возможна дисфункция эндокринной системы. Миопатия Дюшенна поражает дыхательные мышцы, что вызывает пневмонию. Она, в свою очередь, становится причиной летального исхода.
Болезнь Беккера
Такая форма миопатии проявляется у тех пациентов, чей возраст превысил 20 лет. Этот вид заболевания считается легкой формой. Не затрагивается психическое здоровье. Поражаются мышцы таза и бедер. Гипертрофия происходит в мышечной ткани икр.
Форма Эрба
Эта форма имеет и другое название – ювенильная. Ей подвергаются пациенты как мужского, так и женского пола, достигшие 25-и летнего возраста. В некоторых случаях миопатия Эрба возникает и в 20 лет.
Узнаем, почему немеют пальцы левой руки
- Posted on Июнь 5, 2018 Июнь 1, 2018
- Маргарита Юрьевна Новикова
- Невралгия
- Нервно-мышечные заболевания
- 3 просмотра
- 6 минут на чтение
Для такой формы заболевания характерной чертой становится искривление позвоночника, достигаемое путем атрофии мышц таза и туловища. Лопатки становятся похожи на крылья, а при передвижении человек начинает переваливаться. Мышцы в области рта атрофируются. Псевдогипертрофия при форме Эрба в большинстве случаев не проявляется, но бывают и исключения.
Миопатия Эрба, возникшая в раннем возрасте, приводит к полному обездвиживанию. У пациентов, у которых данная форма заболевания проявилась в зрелом возрасте, наблюдается легкое течение.
Миопатия Ландузи-Дежерина
Как и предыдущая форма, Ландузи-Дежерина поражает как мужчин, так и женщин. Заболевание может появиться не только у взрослых, но и у детей от 10-и лет. Сначала миопатия приходится на лицевые мышцы, затем переходит на мускулатуру плеч и груди. Ввиду поражения мышечной ткани, находящейся в области глаз, больные вынуждены спать с открытыми глазами.
Гипертрофия приходится на губы, ограничивая функции мимики. Заболевание Ландузи-Дежерина протекает медленно, но не угрожает жизни больного. Умственные способности не подвергаются негативному воздействию.
Форма Кугельберга-Веландера
Миопатия поражает детей от 2 до 15-и лет. Волна приходится на нижние конечности и таз. В этих участках наблюдается слабость мышц. Это явление имеет прогрессирующий характер, вплоть до невозможности осуществлять движения ногами.
В редких случаях поражаются верхние конечности. Больной испытывает трудности при подъеме со стула, а также при движении по лестнице. Неустойчивость при ходьбе, нередко приводящая к падениям, является характерной чертой данной формы заболевания. Походка больного напоминает «утиный шаг».
Этой форме заболевания подвержены и дети, и взрослые, так как она приходится на возраст от 15 до 30-и лет. Принято считать, что Шарко-Мари возникает у обоих полов, но статистика показывает, что наиболее часто ей подвергаются представители мужского пола.
В мышечной ткани происходят изменения, вызванные расстройствами неврологического характера. Поражение приходится на отдельные группы мышц нижних конечностей.
Ткань может распадаться частично или полностью (второй вариант возможен при поздней стадии). Заболевание воздействует негативно на спинной мозг, ввиду чего больной не может стоять на одном месте. Со стороны такое поведение может показаться странным.
Миопатии делятся на первичные и вторичные. Первичный тип возникает независимо от внешнего воздействия на организм, так как имеет наследственный характер. Если в семье кто-либо из родителей или близких родственников имеет данный диагноз, то высока вероятность, что болезнь передастся новому поколению.
Первичная миопатия может застать человека в возрасте 3-х лет, либо в период полового созревания. Любая форма миопатии, возникшая на фоне плохой наследственности, трудно поддается лечению и имеет тяжелое течение. Заболевание дает о себе знать еще в период грудного вскармливания.
Ребенок не способен кричать громко, как это делают его сверстники. У него преобладают вялые движения. Процесс кормления происходит тяжело, так как ввиду слабых мышц лица не получается крепко обхватить губами сосок.
Вторичная форма миопатии провоцируется другими заболеваниями. Чаще всего причиной становятся:
- Гормональный сбой.
- Тиреотоксикоз (частый процесс синтеза гормонов, вырабатываемых щитовидной железой).
- Склеродермия (сбой коллагена).
- Инфекционное воспаление мышечной ткани (бактериального, вирусного, идиопатического или паразитарного характера).
Нарушение липидного обмена в мышцах, обмен гликогена и метаболизм пуринов также способны стать причиной развития миопатии.
Основная черта большинства форм миопатий – это прогрессирующая слабость, возникающая в мышцах конечностей, а также усталость, возникающая после недолгой физической активности. Слабость с каждым годом увеличивается, перерастая в мышечную атрофию.
Конечности деформируются, ограничивая больных в движении. С трудом получается самостоятельно подниматься со стула, осуществлять движение по лестнице, а также становится невозможно бегать и прыгать.
У больного наблюдаются крыловидно стоящие лопатки. Плечи практически не поднимаются, а живот постоянно выпячен вперед. Человек, страдающий миопатией, передвигается, раскачиваясь из стороны в сторону.
В мышцах конечностей и туловища симметрично происходят патологические изменения. Атрофии больше подвергаются проксимальные отделы рук и ног, что способствует гипертрофированному виду их дистальных участков. Особенно это заметно в области голеней.
Помимо нарастающей слабости, происходит угасание рефлекторных функций сухожилий. Мышечный тонус снижается, приводя к периферическому параличу. Ввиду ограничения телодвижений возникают контрактуры суставов.
Миопатия поражает мышцы лица, что делает невозможными некоторые мимические движения. Больной не может свистеть, хмуриться, улыбаться или вытягивать губы уточкой. Возможно нечеткое произношение гласных звуков, так как заболевание ухудшает работу мышц рта.
Возможно ухудшение функционирования дыхательных мышц. Это приводит к дыхательной недостаточности, а также к застойной пневмонии. В некоторых случаях возникают:
- кардиомиопатия;
- сердечная недостаточность;
- дисфагия мышц глотки и гортани;
- миопатический порез гортани.
При миопатии страдает не только физическая составляющая организма, но и психическая. Больной перестает адекватно рассуждать, его поведение становится беспокойным. Такие перемены характерны не для всех форм заболевания.
Диагностика
Диагностика миопатии осуществляется путем обращения к врачу-неврологу. Основными методами определения заболевания являются:
- ЭНГ (электронейрография).
- ЭМГ (электромиография).
Проведение той, и другой процедуры позволяет не перепутать миопатию с заболеваниями, имеющими схожий характер:
- инфекционная миелопатия;
- нарушение спинномозгового кровообращения;
- опухоль спинного мозга;
- миелит.
При помощи ЭМГ можно зафиксировать изменения, происходящие в мышечной ткани. Наличие большого количества коротких пиков свидетельствует о прогрессирующем процессе, что дает возможность точно поставить данный диагноз.
Диагностика миопатии происходит и путем лабораторных исследований. К примеру, повышенное содержание альдолазы, АСТ, ЛДГ, КФК и прочих ферментов в крови позволяет выявить биохимический анализ. А анализ мочи определяет количество креатина. Если оно увеличено, то это верный показатель миопатии.
Чтобы определить форму заболевания, проводят биопсию мышечной ткани. Это дает возможность установить наличие разбросанных атрофированных миофибрилл. Также можно заметить, как мышечная ткань сменяется на соединительную или жировую. Сопоставление гистохимических, молекулярно-генетических и иммунобиохимических анализов позволяет точно определить заболевание и его форму.
Диагностика поражения сердечной мышцы проводится путем консультации у кардиолога, а также проведения процедур ЭКГ и УЗИ. Если есть симптомы пневмонии, то больного направляют к пульмонологу, а также делают ему рентгенографию легких.
Процесс лечения проходит путем назначения больному глюкокортикостероидных препаратов. Наиболее эффективным считается Преднизолон, суточная доза которого составляет около 100мг. После должного эффекта (появления силы в мышцах), его дозировку снижают до 15мг, но делают это постепенно.
При тяжелых случаях больному назначают метилпреднизолоновую пульс-терапию. Прием гормональных препаратов вызывает множество побочных эффектов, поэтому глюкокортикостероиды некоторым пациентам не выписываются.
В случае бесполезности глюкокортикостероидных препаратов, больному выписывают:
- Метотрексат;
- Циклофосфамид;
- Циклоспорин;
- Азатиоприн.
При форме Шарко-Мари процедура лечения проходит следующим образом:
- назначают витамины В;
- АТФ;
- проводят сеансы физиотерапии;
- делают переливание крови;
- назначают процедуры ЛФК и массаж;
- выписывают антихолинэстеразные препараты.
При свисающих стопах пациенту прописывают ортопедическую обувь. Исключаются физические нагрузки.
Форма Дюшена практически не поддается лечению, так как ее течение прогрессирует, а состояние пациента постоянно ухудшается.
Больному назначают терапию, направленную на поддержание состояния и замедление процесса развития болезни. Она заключается в приеме:
- Витаминов В, Е;
- Оксазила;
- Аминокислот;
- Кальциевых препаратов;
- Галантамина;
- Анаболических гормонов.
Процедура лечения проходит курсами, в условиях постоянного наблюдения у врача. Прием глюкокортикостероидных препаратов позволяет продлить жизнь на несколько лет.
Последствия и осложнения
Ввиду малой подвижности и слабости в мышцах, развивается:
- Дыхательная недостаточность (при поражении дыхательных мышц).
- Сердечная недостаточность.
- Застойная пневмония (ввиду накопления крови в легких).
- Кардиомиопатия.
- Запоры.
- Искривление позвоночника.
Больному трудно передвигаться. Слабость прогрессирует, постепенно переходя в паралич. Становиться трудно жевать и глотать пищу. Возможность летального исхода увеличивается с каждым месяцем.
Возникновение миопатии в младенчестве означает высокую вероятность летального исхода. Если заболевание появилось в более зрелом возрасте, то прогноз может иметь благоприятный характер.
Немалую роль играет развитие болезни и участок, который она охватывает. Если затрагиваются жизненно важные органы (печень, сердце, почки), то заболевание принимает тяжелый характер, что чаще всего приводит к смерти.
Вторичные миопатии имеют более позитивный прогноз, так как своевременное определение и устранение причины, вызвавшей заболевание, способствует регрессу патологии.
Маргарита Юрьевна Новикова
Врач-невролог высшей категории, опыт работы 20 лет. Образование: ЯГМУ
Миопатия Дюшена
Одной из самых грозных первичных мышечных дистрофий, которая начинается в раннем детстве и приводит к летальному исходу до достижения больным 25-летнего возраста, является миопатия Дюшена (полное название – прогрессирующая мышечная дистрофия Дюшена).
Краткая характеристика миопатии Дюшена
Заболевание было впервые описано в 1868 году Дюшеном (Duchenne) и является генетическим. Причем миопатия Дюшена имеет общую, генетически единую форму с миопатией Беккера, однако отличается целым рядом клинических признаков.
Распространению процесса гипотрофии мышц свойственен восходящий характер:
- Сначала в процесс вовлекаются мышцы тазового пояса, а также мышцы проксимальных отделов ног (нижних конечностей),
- Затем страдают мышцы спины и плечевого пояса,
- После этого очередь доходит до проксимальных отделов рук (верхних конечностей).
Уже в самом начале заболевания угасают или значительно снижаются коленные рефлексы, при этом сухожильные рефлексы рук и Ахиллов рефлекс сохраняются ещё очень длительное время.
Другие признаки этого заболевания:
- Прогрессирование кифоза, лордоза или других вторичных деформаций позвоночника,
- Искривление грудной клетки (она становится килевидной или седловидной), стоп,
- Развиваются ретракции сухожилий на фоне контрактур в суставах.
- Очень часто при миопатии Дюшена наблюдаются проблемы с сердцем, а именно кардиомиопатия, симптомами которой становятся аритмия и гипертрофия левого желудочка.
- Хотя обычно при миопатиях интеллект не страдает, в этом случае у 25-30% пациентов наблюдается олигофрения (а именно, в степени дебильности). У остальных больных интеллект сохранен.
Фото миопатии Дюшена:
Миопатия Дюшена является наследственным заболеванием, причем носителями его гена являются представительницы женского пола. Наследуется эта миопатия по рецессивному типу, сцепленному с Х-хромосомой. Более того, около трети всех случаев выявления миопатии Дюшена вызваны новыми мутациями генов. Болеют исключительно мальчики.
Из всего многообразия миопия эта является наиболее злокачественной и быстро прогрессирующей.
Симптомы заболевания
Заболевание начинает проявляться в возрасте 1,5-5 лет, его первыми признаками являются:
- Неустойчивость, двигательная неловкость.
- Постоянные спотыкания и падения во время ходьбы, что развивает у ребенка страх перед ходьбой, чем обусловлена двигательная пассивность.
- Ребенку сложно подниматься по лестнице, а походка становится вразвалочку, «утиной».
- Также затруднительным становится подъем из положения лежа или сидя – ребенок прибегает к так называемым «приемам Гроверса» — это «взбирание по самому себе» и «взбирание лесенкой».
- Яркий признак миопатии Дюшена – мнимая гипертрофия мышц, особенно икроножных: на самом деле происходит не развитие мышц, а их перерождение в жировую и соединительную ткани.
- Как уже упоминалось, одним из симптомов миопатии Дюшена является поражение сердца, причиной которого, по мнению зарубежных исследователей, становится недостаток в кардиомиоцитах дистрофина.
- Наблюдаются признаки миопатии в биоптатах мышц скелета.
- Со временем, по мере прогрессирования мышечной дистрофии развиваются контрактуры крупных суставов, наблюдается эквиноварусная деформация стопы.
- Ближе к 10-12 годам ребенок уже не может передвигаться самостоятельно и вынужден пользоваться инвалидным креслом.
- К 15 годам развивается глубокая инвалидизация больного.
Знаете ли вы, какие бывают симптомы серотонинового синдрома? Причины и последствия.
Всё о принципах лечения миофасциального болевого синдрома можно найти здесь.
Диагностика
Диагностика миодистрофии Дюшена ставится на основании следующих результатов осмотра и анализов:
- На ЭКГ выявляется поражение миокарда латеральной и задне-нижней стенок левого желудочка, что определяется по следующим показателям: высокий зубец наблюдается в отведении V6; глубокий зубец Q наблюдается в отведениях V6, aVF, 2 и 3.
- Также исследуется содержание дистрофина в мышечной ткани (при этом заболевании дистрофия не выявляется).
- В ходе биохимических исследований в плазме крови определяется активность КФК (фермента креатинфорсфокиназы), которая обычно существенно повышена (в том числе у носительниц гена). Иногда для уточнения источника исследуются изоферменты КФК.
- Проводится также генодиагностика.
- Фибрилляции на ЭМГ сообщают о некрозе мышечных волокон.
- Биопсия мышц является одним из основных методов диагностики миопатии Дюшена, причем выбирается умеренно пораженная мышца, поскольку очень ослабленная и существенно пострадавшая мышца окажется неинформативной.
Лечение миопатии Дюшена
При таком тяжелом и быстро прогрессирующем заболевании лечение малоэффективно, обычно в комплексной поддерживающей терапии применяются следующие препараты:
- Группа препаратов, улучшающих обмен веществ в организме: витамины группы В, Е, аминокислоты, препараты кальция, анаболические гормоны, калия оротат.
- Применяется лечение прозерином, галантамином, оксазилом.
- Лечение проводится курсами и, предпочтительнее, в стационаре: ЛФК (особенно замедляющая образование контрактур), так же, как и пассивное растяжение больных мышц, массаж, электрофорез прозерина, лидазы, кальция хлорида, ванны, индуктотермия. Курс лечения повторяется каждые 6-8 недель, а неподвижных детей предпочтительнее лечить на дому.
- Последние годы популярным стало лечение глюкокортикоидами (по схеме через день), которое продлевает жизнь ещё на несколько лет.
Больному миопатией Дюшена нужно обязательно наблюдаться у врача-кардиолога.
Отдельно стоит отметить важность полноценного питания, в котором обязательно должны присутствовать растительные жиры и белки животного происхождения. Стоит избегать крепкого чая, кофе, алкоголя, пряностей, сахара, капусты и картофеля. В рацион должны входить свежие или приготовленные щадящим способом овощи, фрукты, кисло-молочные продукты, овсяная крупа, яйца, мед, морковь, орехи.
Симптомы алкогольной энцефалопатии хорошо освещены в этой статье.
Лечение нейропатической боли у взрослых зачастую представляет собой длительный процесс и требует комплексного подхода. Подробную информацию об этом заболевании можно получить, перейдя по этой ссылке: http://gidmed.com/bolezni-nevrologii/nevralgija/nejropaticheskaya-bol.html.
Профилактика
Профилактика заболевания затруднена ввиду генетических причин его возникновения, вот почему особо важную роль играет генетическое консультирование тех семей, в которых выявлена отягощенная наследственность.
Новейшие методы и разработки молекулярной генетики помогают достоверно определить природу мутации гена, «вычислить» прогноз его заболевания, но самое главное – такие методы позволяют провести перинатальное обследование в случае повторной беременности.
Миопатия Дюшенна: симптомы и лечение
Миопатия Дюшенна — основные симптомы:
- Мышечная слабость
- Задержка речевого развития
- Трудности в ходьбе
- Трудности в обучении
- Сложность при подъеме по ступенькам
- Утиная походка
- Неуклюжесть походки
- Трудности в стоянии
- Опирание на руки при вставании
- Трудности в беге
Миопатия Дюшенна — врожденная патология, которая провоцирует постоянно развивающуюся слабость в мышцах. Болезнь начинает развиваться в детстве. Иногда родители даже не подозревают что ребенок болен: не замечают, что малышу трудно бегать, взбираться по лестнице и даже стоять. Больные с таким диагнозом должны часто проходить обследование. Этот вид миопатии поражает в основном мальчиков, у девочек встречается очень редко (но они носители).
Миопатия Дюшенна — серьезная патология, которая поражает мышцы плеч, туловища и бедер. Руки и пальцы не страдают. Проблемы возникают при ходьбе, беге. Слабость мышц развивается постепенно. В детстве, когда патология начинает развиваться, симптоматика слабая. С возрастом у ребенка усиливается проявление симптомов, качество жизни ухудшается. Патологию диагностируют одному мальчику на 3500 родившихся.
Патогенез заболевания: в мышцах каждого человека есть особый белок под названием дистрофин. У людей, больных миопатией, белка очень мало — из-за этого со временем мышцы начинают слабеть. Причина развития болезни — генетическая предрасположенность: аномальный ген передается от старшего поколения к младшему. Причиной заболевания может стать мутация генов, произошедшая в пубертатном периоде. Тогда у здоровых родителей может родиться ребенок с патологией.
Вероятность того, что мать-носитель передаст сыну дефектный ген, равна 50 %. Если у женщины родится дочь, с той же вероятностью девочка может стать носителем.
Заболевание генетического характера, когда в организме действует ген Дюшенна.
Существуют определенные факторы, которые могут оказать влияние на развитие патологии:
- генетическая предрасположенность;
- брак между близкими родственниками;
- наследование патологии от матери (встречается чаще всего);
- мутация генов при внутриутробном развитии;
- аномалия структуры хромосомы;
- сильное поражение дистрофина;
- патологические биохимические процессы;
- замещение соединительной или жировой тканью мышечных волокон.
Миопатия Дюшенна развивается вследствие системных заболеваний соединительной ткани.
Симптоматика
Симптомы болезни Дюшенна начинают проявляться уже в трехлетнем возрасте. В этот период возникают признаки, на которые родители должны обратить внимание:
- малышу тяжело ходить, стоять, бегать, подниматься по ступенькам, походка отличается неуверенностью, ребенок ходит вразвалочку;
- ребенок немного постарше опирается на руки, чтоб встать;
- чадо отстает в обучении по сравнению со сверстниками.
В некоторых случаях выявить миопатию можно по такому признаку: у ребенка плохо развивается речь. Диагностика заболевания обычно не представляет сложности, ибо у миопатии Дюшенна симптомы весьма специфические.
Диагностика
При обращении к доктору первое, что врач сделает, — проведет наблюдение. Педиатр посмотрит, как ребенок ходит, бегает, встает с пола. Если у медицинского специалиста возникнут подозрения на развитие миопатии, будет назначен специальный анализ крови.
Тесты для диагностики болезни Дюшенна:
- Анализ на креатинкиназу (фермент). Если его уровень во много раз превышает норму, ставится соответствующий диагноз. Если показатель в норме, миопатия Дюшенна исключается — врач ищет другие причины патологического состояния.
- Проводится биопсия мышечной ткани. Проводят манипуляцию под общим наркозом — берут у ребенка небольшой фрагмент мышечных волокон для дальнейшего исследования. Благодаря анализу уточняют уровень дистрофина и состояние мышечной ткани.
- Генетическое тестирование. Для этого проводится забор крови. В дальнейшем выявляют гены, которые повлияли на развитие врожденной патологии.
Диагностируют болезнь в основном с помощью последнего метода.
На сегодняшний день миодистрофия Дюшенна не лечится. Есть определенная терапия, помогающая облегчить состояние больного. Процесс развития болезни при соблюдении медикаментозного курса в дальнейшем немного замедляется.
Метод лечения миопатии Дюшенна подбирается в зависимости от возрастной группы. Бывают случаи, когда следует совмещать несколько способов терапии.
Больным в младшем дошкольном возрасте обычно не требуется особого лечения. Врач может порекомендовать родителям следующее:
- консультация, как правильно давать физические нагрузки больному ребенку;
- консультация генетика — если родители хотят знать, являются ли они носителями (это необходимо сделать тем, кто собираются еще рожать детей).
В этом возрасте ребенка необходимо постоянно обследовать у специалиста, что поможет начать своевременное лечение.
Способы терапии детей в возрасте от 5 до 8 лет:
- ребенку рекомендуют применять поддержку для ножных мышц — шину для лодыжки, надеваемую только на ночь, либо длинные шины для голени;
- назначают применение кортикостероидов — медикаменты немного замедлят развитие заболевания, мышцы будут сильными, но такие препараты вызывают серьезные осложнения, поэтому ребенок должен находится под присмотром доктора.
После 8 лет мышцы ребенка, у которого миодистрофия Дюшенна, сильно слабеют. Ему трудно ходить, чадо начинает передвигаться на инвалидной коляске (обычно с 9 до 11 лет). Пациенты, которые рано начали принимать лекарственные препараты, ходят немного дольше.
После того как ребенок уже не встает с инвалидного кресла, развиваются другие патологии. В это время посещения доктора становятся чаще. Чем раньше будет начато лечение выявленного заболевания, тем положительнее будет результат.
Родители в этот период должны помогать ребенку передвигаться в кресле и обустроить для комфортного передвижения в инвалидной коляске дом.
По мере того как ребенок взрослеет, слабость мышц доставляет все больший дискомфорт. Человеку чаще необходима помощь родных, возникают легочные инфекции.
Возможные осложнения
Наиболее серьезное осложнение — сокращение срока жизни. Из-за быстрого развития заболевания мышцы слабеют, появляются серьезные заболевания сердца. Работа дыхательной системы нарушается.
Если миодистрофия типа Дюшенна Беккера обнаружена на ранних стадиях развития, а способ терапии подобран правильно, больной может прожить до 30 лет.
Параллельно с этим заболеванием развиваются и осложнения:
- остеопороз;
- болезни позвоночника и суставов;
- проблемы с желудочно-кишечным трактом;
- нарушения в работе дыхательной системы;
- сердечно-сосудистые заболевания.
Профилактика
Профилактических мер не существует, потому что болезнь передается по наследству.
Будущим родителям перед планированием беременности необходимо проконсультироваться с врачом-генетиком. Особенно это касается тех пар, у которых в анамнезе отмечены случаи подобных заболеваний.
Родителям нужно внимательно относиться к ребенку и при первых симптомах, которые свидетельствуют о развитии слабости мышц, незамедлительно обращаться за медицинской помощью в поликлинику по месту жительства.
Для максимального продления жизни пациента с болезнью Дюшенна нужно обязательно в полной мере выполнять все рекомендации врачей, систематически проходить медицинское обследование и укреплять иммунную систему.
Если Вы считаете, что у вас Миопатия Дюшенна и характерные для этого заболевания симптомы, то вам могут помочь врачи: педиатр, терапевт, генетик.
Также предлагаем воспользоваться нашим сервисом диагностики заболеваний онлайн, который на основе введенных симптомов подбирает вероятные заболевания.
Остеомаляция — это заболевание, которое начинает прогрессировать вследствие нарушения минерализации костных тканей. Как результат, происходит патологическое размягчение костей. Недуг по своей этиологии и клинике напоминает такую болезнь, как рахит у детей, который происходит из-за гиповитаминоза D3.
Гиперпаратиреоз – хроническая патология околощитовидных желез, прогрессирующая вследствие возникновения опухолей или усиленного разрастания их тканей. Патология характеризуется повышенным продуцированием паратгормона, влияющего на кальциевый обмен. Избыточное его содержание в крови становится причиной того, что из костей вымывается кальций, а это, в свою очередь, приводит к тяжёлым осложнениям.
Спинальная амиотрофия Верднига-Гоффмана — генетическая патология нервной системы, при которой проявляется мышечная слабость по всему организму. Такое заболевание нарушает способность человека сидеть, самостоятельно передвигаться и ухаживать за собой. В современном мире пока нет эффективной терапии, которая дала бы положительный результат.
Рак позвоночника может быть актуальным заболеванием, проявляющимся в форме первичной либо вторичной злокачественной опухоли области позвоночника. Опухоли вторичные – это метастазы новообразований злокачественного типа, чье развитие изначально происходит в каком-либо из органов (предстательная железа, легкие, желудок, грудная полость и т.д.). Рак позвоночника, симптомы которого заключаются в болевых ощущениях, развивается постепенно, в течение нескольких недель/месяцев, после чего отмечается усиление этих проявлений.
Миопатия – врождённая патология, причиной которой являются определённые мутации в генах. Механизм развития болезни окончательно не изучен, поэтому врачи не могут точно определить, когда у пары может родиться больной ребёнок. Бывает и так, что у совершенно здоровых отца и матери может появиться ребёнок с любой формой миопатии. Вообще же болезнь связана с нарушениями обменных процессов в тканях мышц, которые из-за этого теряют креатин, что приводит к их дистрофии.
При помощи физических упражнений и воздержанности большая часть людей может обойтись без медицины.
Особенности развития и лечения миопатии Дюшена
Миопатия Дюшена представляет собой одну из самых тяжелых форм первичной мышечной дистрофии. Это врожденное заболевание. Оно проявляется в раннем детстве, характеризуется тенденцией к быстрому прогрессированию и в большинстве случаев приводит к летальному исходу еще до достижения пациентом 25-летнего возраста.
Отличительной чертой патологии считается то, что страдают от нее преимущественно мальчики, а девочки, за редким исключением, являются лишь носителями дефектного гена, передающегося наследственным путем.
Почему возникает патология?
Представленный вид мышечной дистрофии является заболеванием наследственного характера, которое возникает и развивается из-за действия так называемого гена Дюшена. Специалисты выделяют следующие причины, которые могут спровоцировать появление данной патологии:
- Генетический фактор.
- Браки между близкими родственниками.
- Наследование заболевания по материнской линии (наблюдается в большинстве случаев).
- Последствие так называемой новой мутации (встречается достаточно редко, приблизительно в 30% случаев).
- Структурная хромосомная аномалия.
- Глобальное поражение особого гена, ответственного за выработку белка (дистрофина).
- Патология биохимических процессов.
- Нарушение выработки энзимов в организме человека.
- Замещение мышечных волокон жировыми или же соединительными тканями.
Как проявляется заболевание?
Первые признаки прогрессирующей мышечной дистрофии Дюшена проявляются уже в самом раннем детстве и становятся заметны при достижении ребенком полутора лет. Малыши, пораженные этим недугом, значительно отстают в психомоторном развитии от своих сверстников. Они длительное время не могут самостоятельно встать на ножки, гораздо позже остальных детей начинают ползать, вставать и садиться.Больной мышечной дистрофией ребенок редко начинает ходить к положенному времени, а впоследствии часто падает и передвигается с большим трудом. Типичным признаком считается постоянное опирание (облокачивание) на руки при попытках малыша подняться с места. Кроме того, для данного вида патологии характерны следующие симптомы:
- Некроз волокон.
- Перерождение и замещение мышечных тканей.
- Симметричная атрофия мышц.
- Псевдогипертрофия икроножных мышц, сопровождаемая увеличением соединительных тканей и жировых отложений.
- Патологическая утомляемость во время физических нагрузок.
- Развитие лордоза.
- Снижение общего тонуса проксимальных мышечных групп.
- Изменение и постепенное исчезновение мышечных рефлексов.
- Развитие сопутствующих сердечно-сосудистых заболеваний.
- Поражение дистальной мускулатуры (проявляется на поздних стадиях заболевания).
- Нарушения нейроэндокринного характера.
- Деформация позвоночного столба.
- Проявление синдрома Иценко-Кушинга.
- Патологическое деформирование стоп и области грудной клетки. Неустойчивость походки.
- Адипозо-генитальная дистрофия.
- Сужение в костномозговом канале.
- Психические расстройства.
- Утрата сухожильных рефлексов.
- Олигофрения и замедленное речевое развитие.
- Нарушение координации движений.
- Развитие так называемой «утиной» походки и суставные контрактуры.
- Кардиомиопатия и сердечная дистрофия.
- Нарушения моторики желудочно-кишечного тракта.
- Сколиоз и костные деформации.
- Вегетативные расстройства и поражение дыхательных мышц.
- Развитие эндокринных заболеваний.
Ярко выраженные при миопатии Дюшена симптомы в виде интеллектуальных расстройств и поражения внутренних органов пациента проявляются несколько позднее патологических мышечных признаков. В большинстве случаев это происходит при достижении ребенком 8 — 10 лет.
В чем опасность патологии?
Прогрессирующая дистрофия Дюшена считается одним из самых тяжелых и опасных видов патологий. Развивается заболевание постепенно. В первую очередь происходит атрофия ягодичных и тазобедренных мышечных групп. Ребенку тяжело вставать с места, подниматься по лестнице, функции опорно-двигательного аппарата заметно ограничены. Постепенно атрофируются группы мышц верхней части корпуса, а затем и все остальные мышцы.
Ближе к 5-летнему возрасту развивается псевдогипертрофия икр: они кажутся непропорционально увеличенными за счет накопления жировых отложений и интенсивного разрастания соединительных тканей. Ребенок страдает патологической утомляемостью при минимальных физических нагрузках, развивается мышечная слабость, которая не проходит даже в состоянии покоя. Костные деформации, характерные для данного вида миопатии, приводят к внешним изменениям: появлению так называемой осиной талии и крыловидных лопаток.
Миопатия Дюшена развивается стремительно: уже к 10-летнему возрасту маленький пациент превращается в полного инвалида, неспособного к самостоятельному передвижению. Также наблюдаются задержки умственного развития, проявляющиеся в имбецильности или же дебильности, характерных для олигофрении. Однако основная опасность миопатии Дюшена заключается в сопутствующих патологических процессах, происходящих во внутренних органах. В первую очередь поражается сердечно-сосудистая и дыхательная системы. Именно осложнения в виде пневмонии, сердечной недостаточности, заболеваний респираторной системы и т. п. становятся причиной гибели пациента.
Каковы способы диагностики?
Помимо изучения общей клинической картины и характерных симптомов, для диагностирования прогрессирующей дистрофии Дюшена пациенту назначают ряд исследований. К ним относятся следующие процедуры:
- Электромиография, позволяющая определить показатели скорости нервных импульсов в мышечных группах.
- Развернутый анализ крови для выявления биохимических показателей, в частности уровня фермента креатинфосфокиназа, значительно повышенного в случае миопатии.
- Мышечная биопсия.
- Магниторезонансная томография.
- Проведение специального генетического тестирования.
- Электрокардиограмма.
Следует подчеркнуть, что своевременная диагностика предоставляет возможность выявить в организме дефектный ген Дюшена и начать лечение, что позволит избежать ряда тяжелых осложнений и значительно улучшить качество жизни маленького пациента.
Как лечится патология?
На сегодняшний день полностью излечить данное заболевание невозможно, однако существует множество способов, позволяющих облегчить состояние больного и увеличить продолжительность его жизни.
- Ортопедическое вмешательство.
- Массажи.
- Физиотерапия.
- Медикаментозное лечение, заключающееся в приеме Преднизолона, способствующего увеличению мышечной массы и заметно замедляющего процесс развития патологии. Кроме того, пациентам с данным диагнозом показаны анаболики: Прозерин, Калия оротат, Галантамин, препараты кальция, разного вида аминокислоты, а также глюкокортикостероиды.
- Лечебная физкультура.
- Использование специальных ортопедических протезов.
- Генная терапия, представляющая собой искусственное введение в организм пациента двух видов генов: утрофина и дистрофина.
- В некоторых случаях при развитии контрактур, а также для фиксирования суставов применяется хирургическое лечение.
- Витаминная терапия.
- Электрофорез.
- Индуктотерапия.
Помимо этого, очень важно обеспечить ребенку правильное питание. Ежедневный рацион маленького пациента непременно должен включать в себя свежие овощи и фрукты, яйца, мед, злаковые, молочные продукты, богатые кальцием. А вот чай, кофе, специи, сладости, острая и жирная пища больным, страдающим мышечными патологиями, категорически противопоказаны.
Миопатия Дюшена — тяжелая мышечная патология, зачастую приводящая к гибели пациента в молодом возрасте. Однако благодаря достижениям современной медицины при своевременном грамотном лечении можно избежать ряда осложнений и значительно увеличить продолжительность жизни больного.
Статья написана по материалам сайтов: www.krasotaimedicina.ru, nevralgia.ru, gidmed.com, simptomer.ru, nervzdorov.ru.
»