Прогрессирующая оссифицирующая фибродисплазия: лечение
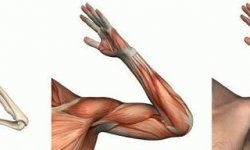
Прогрессирующая оссифицирующая фибродисплазия – тяжелое генетическое заболевание, характеризующееся нарушением обмена в тканях мышц, сухожилий и связок, что приводит к образованию в них кальцинатов. Симптомами этого состояния являются уплотнения в мягких тканях шеи, спины и конечностей, мышечная гипотония, в дальнейшем – выраженные деформации позвоночника, скованность движений вплоть до инвалидизации больных. Диагностика прогрессирующей оссифицирующей фибродисплазии производится на основании данных настоящего статуса пациента, рентгенологических и молекулярно-генетических исследований. Специфического лечения патологии на сегодняшний день не существует, но имеются обнадеживающие результаты опытов на клеточных культурах больных данным заболеванием.
Содержание
- Прогрессирующая оссифицирующая фибродисплазия
- Причины прогрессирующей оссифицирующей фибродисплазии
- Симптомы прогрессирующей оссифицирующей фибродисплазии
- Диагностика и лечение прогрессирующей оссифицирующей фибродисплазии
- Фибродисплазия
- Историческая справка
- Клинические проявления фибродисплазии
- Причины развития заболевания
- Диагностика фибродисплазии
- Стадии окостенения на рентгеновских снимках
- Дополнительные аномалии развития скелета
- Факторы развития фибродисплазии
- Стадии развития оссифицирующей прогрессирующей фибродисплазии
- Поэтапное развитие болезни
- Лечение оссифицирующей прогрессирующей фибродисплазии
- Профилактические мероприятия для улучшения качества жизни у больных с фибродисплазией
- Болезнь, приводящая к инвалидности — фибродисплазия (болезнь Мюнхеймера)
- Что такое фибродисплазия?
- Симптоматика
- Диагностика и лечение
- Прогрессирующая оссифицирующая фибродисплазия
- Прогрессирующая оссифицирующая фибродисплазия (ФОП)
- Характеристика заболевания
- Как развивается
- Диагностика
Прогрессирующая оссифицирующая фибродисплазия
Причины прогрессирующей оссифицирующей фибродисплазии
Этиология прогрессирующей оссифицирующей фибродисплазии заключается в нарушении регуляции процессов формирования костной ткани. В эмбриональном периоде за развитие костей отвечает ряд рецепторов, работа которых приводит к образованию твердой соединительной ткани в соответствующих областях тела. При развитии этого заболевания возникает мутация гена ACVR1, расположенного на 2 хромосоме. Продуктом экспрессии данного гена является специфический белок – активин-рецептор 1 типа. Он относится к более обширной группе BMP-рецепторов, которые обладают способностью связывать вещества-факторы роста костной ткани и передавать соответствующие сигналы внутрь клеток. Чаще всего причиной прогрессирующей оссифицирующей фибродисплазии являются миссенс-мутации (например, Arg206His) в 6 экзоне гена ACVR1.
В результате этого генетического нарушения происходит образование дефектной формы активина-рецептора 1 типа, который начинает образовываться не только в эмбриональном периоде и в остеобластах у взрослого человека, но и в клетках волокнистой соединительной ткани. Дальнейший патогенез прогрессирующей оссифицирующей фибродисплазии изучен недостаточно по причине редкости этого состояния. Однако большинство исследователей склоняются к мнению, что дефектный рецептор становится способным реагировать не только на соединения из группы BMP (bone morphogenetic protein – белки костного морфогенеза), но и на другие вещества – в частности, факторы воспаления. Доказательством этой теории служит тот факт, что очаги гетеротопической оссификации чаще всего возникают на месте повреждения мягких тканей – ушибов, порезов, укусов насекомых. Кроме того, при гистологическом изучении патологических уплотнений в них всегда наблюдается умеренная лимфоцитарная инфильтрация, что указывает на воспалительный или иммунный характер процесса.
Мутации гена ACVR1 приводят не только к аномальному костному морфогенезу у детей и взрослых, они также нередко обуславливают ряд врожденных пороков развития опорно-двигательного аппарата. Наиболее распространенными проявлениями прогрессирующей оссифицирующей фибродисплазии такого типа являются микродактилии, варусное искривление большого пальца стопы (клинодактилия). Из других, более редких костных аномалий, отмечают сращение тел шейных позвонков, дисплазии метафизов, сращение реберно-позвоночных суставов. По причине сочетания дисплазии костной ткани и слабости мышечного корсета спины у больных часто развиваются тяжелые искривления позвоночника, что приводит к вторичным неврологическим нарушениям.
Симптомы прогрессирующей оссифицирующей фибродисплазии
При рождении прогрессирующая оссифицирующая фибродисплазия может проявлять себя лишь нарушениями формирования костей пальцев рук и ног, никаких других изменений при этом не обнаруживается. Начало заболевания внезапное, может произойти без всяких видимых причин в возрасте от нескольких месяцев до 3-5 лет. Обычно первыми симптомами данной патологии становятся уплотнения в мышечных тканях шеи, спины и рук, размеры которых могут составлять от 1 до 10 сантиметров. При пальпации очаги бывают болезненными, кожа над ними либо не изменена, либо (в редких случаях) гиперемирована. При прогрессирующей оссифицирующей фибродисплазии такие уплотнения могут появляться на месте ушибов, порезов и других травм мягких тканей.
Походка больных прогрессирующей оссифицирующей фибродисплазией, как правило, характеризуется скованностью, малыми шагами. Лицо амимичное на фоне напряженных мышц шеи и спины. По мере нарастания количества и размеров уплотнений объем движений в суставах постепенно уменьшается, приводя на терминальных этапах заболевания практически к полному обездвиживанию. Нередко отмечаются боли неврологического характера, обусловленные сдавлением корешков спинномозговых нервов, также могут поступать жалобы на нарушение кожной чувствительности. Все эти проявления прогрессирующей оссифицирующей фибродисплазии постепенно нарастают и становятся все более выраженными.
Описаны случаи, когда очаги на первоначальном этапе развития самопроизвольно рассасываются (в том числе и под воздействием некоторых лекарственных средств), однако в последующем уплотнения неизменно возникают снова. В течение 2-3 недель происходит оссификация, после чего их самопроизвольное или терапевтическое разрешение становится невозможным. У взрослых больных прогрессирующей оссифицирующей фибродисплазией выявляются десятки таких очагов, которые могут сливаться между собой, образуя картину «второго скелета». Это обстоятельство наряду с многочисленными деформациями костей приводит к глубокой инвалидизации больных и нередко провоцирует развитие многочисленных нарушений со стороны внутренних органов с последующим летальным исходом.
Диагностика и лечение прогрессирующей оссифицирующей фибродисплазии
Рентгенологическое исследование на ранних этапах заболевания может обнаруживать врожденные пороки развития опорно-двигательного аппарата – клинодактилию больших пальцев ног, дисплазию метафизов, укорочение длинных трубчатых костей конечностей. По мере развития гетеротопической оссификации в сухожилиях, фасциях, межмышечных соединительнотканных перегородках выявляются сначала одиночные, а затем и множественные тени, имеющие костную плотность. В далеко зашедших случаях прогрессирующей оссифицирующей фибродисплазии тени сливаются между собой и нередко затрудняют визуализацию внутренних органов и других глубоко расположенных структур. При помощи методов современной генетики можно диагностировать это состояние путем поиска мутаций в гене ACVR1.
Специфического лечения прогрессирующей оссифицирующей фибродисплазии не существует, а паллиативная терапия резко ограничена в своих возможностях по причине детского возраста большинства пациентов и специфической реакции организма. Многочисленные попытки устранить очаги гетеротопической оссификации хирургическим путем заканчивались неудачно – после операции из-за повреждения тканей рост уплотнений резко усиливался. На ранних этапах заболевания немного затормозить развитие патологических очагов можно при помощи высоких доз кортикостероидных препаратов и других средств, угнетающих воспалительные процессы (НПВС, ингибиторы лейкотриенов, блокаторы мастоцитов). В последние годы получены первые лабораторные данные по поводу специфического лечения прогрессирующей оссифицирующей фибродисплазии – при помощи особой РНК удалось ингибировать экспрессию дефектного гена ACVR1, не затрагивая его здоровую гомологичную копию в стволовых клетках больных. Возможно, в дальнейшем это позволит с успехом лечить пациентов, страдающих данным заболеванием, или, как минимум, значительно повысить их качество жизни.
Прогноз и профилактика прогрессирующей оссифицирующей фибродисплазии
Фибродисплазия
Фибродисплазия или болезнь Мюнхеймера (от лат. fibro — волокно, dis — расстройство, нарушение, plasis — строение, структура и os, ossis — кость, facio — делать; окостенение) — это патологическое состояние организма, среди всего населения планеты встречается очень редко, характерной чертой данного заболевания является перерождение мягкой скелетной ткани в костную.
Историческая справка
Впервые про оссифицирующую прогрессирующую фибродисплазию было упомянуто в 1648 году, когда один из докторов описал этот феномен, так как у него было так называемая «окаменевшая» пациентка. Более детально болезнь была описана уже в 1869 году немецким врачом, он не обнаружил связь между половыми признаками и расовой принадлежностью пациента, а также этнической предрасположенностью.
Патология начинает развиваться в детском возрасте, прогрессировать фибродисплазия продолжает в возрасте от 3-х до 4-х лет. Также ученым известны случаи, когда болезнь начала себя проявлять в трехмесячном возрасте.
Клинические проявления фибродисплазии
На данный момент существует только два клинических проявления, после которых можно поставить диагноз оссифицирующая прогрессирующая фибродисплазия. К ним относят: аномалия развития большого пальца, которая наблюдается с рождения ребенка и прогрессирующие гетеротопические окостенения на разнообразных участках тела.
Маленькие пациенты с оссифицирующей фибродисплазией практически ничем не выделяются среди здоровых детей, отличием являются только аномалия большого пальца, которую можно наблюдать у всех детей с данным диагнозом. В течение первых 10 лет жизни ребенка у него начинают развиваться отек мягких тканей, который по своей структуре очень болезненный и воспален. Позже именно этот процесс подвергает трансформации мягкие ткани, перерождая их в гетеротопические кости. Впоследствии именно гетеротопические кости начинают замещать соединительную ткань, а также скелетные мышцы, что в свою очередь приводит к формированию так называемого «второго» скелета, и в конечном итоге человек может быть полностью обездвижен.
Хирургическим путем нельзя удалить гетеротопические кости, так как это приводит большей травматизации близлежащих тканей с последующим развитием новых участков окостенения. Сначала заболевание начинает поражать отделы позвоночника, после переключается на участки черепа и остальные области тела. Но есть мышечные структуры, которые не поддаются влиянию фибродисплазии, к таким структурам следует отнести: сердечную мышцу, диафрагму и язык.
Костные участки формируются поэтапно и впоследствии наносят необратимый вред здоровью человека. Пациенты с данной патологией в возрасте 30-ти лет уже вынуждены передвигаться на инвалидной коляске, и выполнять повседневные дела при помощи своих родных и близких. Быстрая потеря в весе может привести к обездвиженности в области суставов, а также в результате образования костной ткани на стенках грудной клетки приводит к развитию сердечной недостаточности. Такие пациенты живут примерно до 40-ка лет. Умирают они в основном от синдрома грудной недостаточности.
Причины развития заболевания
Причина, по которой развивается данное заболевание, до сих пор точно не установлена. Ученые выяснили, что данная патология передается по наследству, а именно аутосомно-доминантным путем. Имеется в виду, что пациент с фибродисплазией в 100 процентах случаев будет иметь потомство с такой же патологией.
Ученые подтвердили, что во время блокировки мутировавшего гена, возможно притормозить развитие неизлечимого заболевания. Обычно люди с фибродисплазией не доживают до репродуктивного возраста, и поэтому у них не рождаются дети, а также в виду наличия патологии просто не могут иметь детей. Женщинам с фибродисплазией очень тяжело решиться на рождение ребенка, поскольку роды могут быть для них смертельными. Есть пациенты, которые являются только носителями данного гена и при воздействии определенных факторов болезнь может прогрессировать. К таким факторам следует отнести:
- травмирование кожных покровов;
- оперативное вмешательство;
- введение вакцины в организм человека;
- воздействие вирусных инфекций.
Диагностика фибродисплазии
Очень часто специалисты не могут поставить правильный диагноз. Поскольку не все специалисты обращают внимание на активно формирующиеся отеки мягких тканей в области шеи и головы, а также верхней части спины, наличие аномально развитого большого пальца ноги. Очень редко только по одним клиническим проявлением можно диагностировать болезнь, обычно для этого нужно пройти рентгенографию, на которой будут обнаружены участки окостенения. Фибродисплазию чаще всего путают с саркомой мягких тканей, лимфедемой или агрессивным фиброматозом. При проведении диагностики у маленьких детей часто проводятся ненужные анализы, а именно биопсия, которая дополнительно травмирует мягкие ткани и приводит к образованию окостеневших участков.
Стадии окостенения на рентгеновских снимках
При помощи рентгенографии можно определить стадию окостенения у пациента, они делятся на:
- Первая стадия (инфильтрация) характеризуется разрастанием молодой дегенеративной ткани, и образования вторичных дегенеративных изменений в мышечных структурах. На рентгеновских снимках эти изменения малозаметны.
- Вторая стадия или фиброзная индурация. Происходит рубцевание молодой ткани и развивается вторичная атрофия мышечных структур. На рентгеновских снимках можно увидеть нежные тени, напоминающие собой костную мозоль.
- Третья стадия или собственно окостенение. Это формирование собственно костной ткани на участках повреждения мягких тканей. На рентгеновских снимках видно очень четко участки с окостенением, а именно определяются крупные участки окостенения в мышечных тканях, которые срощены с костями, позвоночный столб похож на бамбуковую трость, и определяются анкилозы суставов.
Дополнительные аномалии развития скелета
Помимо больших пальцев аномальному развитию подвергается шейный отдел позвоночника. Жесткость в области шеи являются ранним диагностическим признаком фибродисплазии у пациентов. Дополнительно специалисты отмечают такие патологии: изогнутые, и в тоже время короткие пальцы, а также широкие кости бедер. Аномалии развития бедренных костей определяются на генетическом уровне.
Факторы развития фибродисплазии
Стадии развития оссифицирующей прогрессирующей фибродисплазии
Выделяют несколько стадий развития процесса окостенения:
- Развитие небольшого участка воспалительного процесса, который ограничен в размерах.
- Прогрессирование болезни и задействование в воспалительный процесс близлежащих тканей.
- Образование бессосудистого уплотнения.
- Формирование хряща.
- Собственно окостенение.
Поэтапное развитие болезни
В зависимости от стадии, возраста и воздействия внешних факторов болезнь может вести себя по-разному. После завершения стадии обострения на протяжении определённого промежутка времени, если пациент не подвергался травмированию, фибродисплазия может не проявлять себя. Но обычно она активно прогрессирует, и в результате человек быстро теряет свою подвижность и становится прикованным к инвалидному креслу. Сначала формируются контрактуры в области локтевых и коленных суставов, что в свою очередь начинает ограничивать человека в движении. Далее подвергаются поражению более мелкие суставы и хрящи, которые соединяют ребра с позвоночником. Период, когда начинается стадия обострение заболевания невозможно предугадать, поскольку процесс дисплазии обычно запускает иммунная система. Практически все мышечные структуры организма подвержены окостенению.
Лечение оссифицирующей прогрессирующей фибродисплазии
Современная медицина еще не изобрела лекарство, которое лечило бы фибродисплазию. Но и методы по облегчению состояния больного значительных результатов не приносят. При первых проявлениях заболевания положительная динамика наблюдается во время использования глюкокортикостероидов, тем самым уменьшая проявления болезни и улучшая качество жизни пациента, но к полному выздоровлению не приводит. Полностью избавиться от костных наростов при помощи глюкокортикостероидов не представляется возможным.
Период обострения болезни начинается очень резко, даже спонтанно, и также неожиданно заканчивается, оставляя за собой костное образование. В современном мире тратится очень большое количество времени и средств на изучение данной патологии, целью данных исследований является улучшение качество жизни и состояния здоровья таких пациентов. Было проведено исследование на лабораторных мышах, при использовании лекарственного препарата было установлено, что он помогает бороться с признаками фибродисплазия вызванной как мутировавшим геном, так и травмами. На людях, к сожалению, эти испытания не проводились, потому что невозможно найти достаточное количество пациентов с фибродисплазией для проведения клинических испытаний. В виду отсутствия пациентов, данный лекарственный препарат будет находиться на стадии разработки еще длительное время.
Но в 2014 вроде удалось собрать необходимое количество пациентов для клинических испытаний лекарственного препарата «Паловаротен», испытания проводятся сразу в нескольких городах – Париж, Филадельфия и Сан-Франциско.
Среди клиник по лечению оссифицирующей прогрессирующей фибродисплазии самой лучшей считается лаборатория Маккея при Пенсильванском Федеральном Университете, которой руководит доктор медицинских наук Фредерик Каплан. Это единственная в мире лаборатория, которая специализируется на изучение феномена фибродисплазии.
В России также можно провести диагностические процедуры относительно фибродисплазии, которые проводятся в медико-генетическом научном центре РАМН в Москве по ул. Москворечье д. 1. В НИИ Ревматологии профессор Никишина И.П. и в ММА имени Сеченова профессор Голованова Н.Ю. (Москва) проводят терапевтическое лечение, а также в Иркутском государственном медицинском университете доктор медицинских наук Калягин А.Н. (Иркутск) и в Казанской Государственной Медицинской Академии доктор медицинских наук Мальцев С.В. (Казань).
Профилактические мероприятия для улучшения качества жизни у больных с фибродисплазией
При соблюдении некоторых простых правил можно избежать быстрого прогрессирования болезни. Для этого необходимо избегать больных с ОРВИ, травмирования тканей, перегревания, а также переохлаждения организма. Это не гарантирует полную защиту от прогрессирования болезни, но приостановит ее на некоторое время. Своевременная диагностика и правильная постановка диагноза позволяет начать раннюю терапии глюкокортикостероидами, и тем самым продлить жизнь пациенту и улучшить качество его жизни.
Также в профилактических целях можно использовать Интерферон человеческий лейкоцитарный. Во время длительной терапии интерфероном понижается количество мышечных и подкожных инфильтратов. Также возможно использование биофосфонатов и этилендиаминтетрауксусной кислоты.
К сожалению, исход данного заболевания является не очень благоприятным для пациента. Продолжительность жизни таких пациентов очень разнообразна. Все зависит от воздействия внешних факторов и генетических модификаций.
Болезнь, приводящая к инвалидности — фибродисплазия (болезнь Мюнхеймера)
В этой статье мы расскажем вам о тяжелейшем инвалидизирующем заболевании, которое встречается очень редко — 1 человек на 2 млн населения. Всего в мире насчитывается около 500 больных этим недугом. Он заключается в том, что мышцы больных постепенно превращаются в кость, а сам человек постепенно, по сути, становится статуей и утрачивает возможность двигаться. Название этого недуга — фибродисплазия.
Давайте узнаем, из-за чего развивается эта болезнь, кто ей подвержен и реально ли ее вылечить.
Что такое фибродисплазия?
Это болезнь, спровоцированная изменением гена ACVR1, которая проявляется наследственными дефектами развития. Этот недуг получил также название «болезнь второго скелета». Прогрессирующая оссифицирующая фибродисплазия (ПОФ, болезнь Мюнхеймера) провоцируется воспалениями в сухожилиях, мышцах, связках, мягких тканях, которые с течением времени костенеют. Говоря проще, ушибы и даже небольшие ранки не поддаются лечению и на их месте появляются костные образования, по этой причине недуг получил еще одно название «болезнь второго скелета».
У новорожденных можно с 95% вероятностью определить ПОФ — большой палец ноги загнут внутрь и зачастую там отсутствует сустав. Обычно болезнь проявляет себя в течение первых 10 лет жизни ребенка и совершенно внезапно. У детей все начинается с шеи и постепенно спускается до нижних конечностей.
Скорость развития также невозможно установить точно, но чем больше травм получает больной и чем больше хирургических вмешательств происходит, тем быстрее развивается недуг.
Довольно часто ПОФ путают с раком и прочими болезнями, в результате которых могут развиться подобные костные образования. Врачи удаляют эти образования, но, к сожалению, это приводит к усугублению ситуации и ускорению развития фибродисплазии.
В этой передаче рассказывается о двух больных девушках (русской и американки). Они рассказывают о том, как все началось и возможно ли вылечить ПОФ. У американской девушки врачи первоначально диагностировали рак и ампутировали больную руку в раннем детстве, и только потом выяснилось, чем она в действительности больна.
Что именно запускает мутацию на генном уровне, ученые пока не могут выяснить. На данный момент известно только то, что это наследственный недуг и передается аутосомно-доминантным путем. Это значит, что у больного ПОВ 100% будут дети с точно такой же патологией. Однако люди с болезнью Мюнхеймера редко доживают до возраста, когда могут рожать детей. А если и доживают, то женщинам смертельно опасно беременеть из-за изменений, происходящих в организме. Так что ПОФ является итогом индивидуальных генетических мутаций.
Только в 2006 году ученые выявили, мутация какого именно гена причастна к развитию фибродисплазии и сейчас проходят исследования, чтобы создать препарат, позволяющий лечить этот недуг.
Зачастую у носителей мутировавшего гена недуг никак не показывает себя до какого-то времени. Толчком к развитию могут послужить:
- травмы,
- вакцинации,
- вирусное заражение,
- хирургические вмешательства.
Симптоматика
Самыми очевидными симптомами болезни Мюнхеймера является окостенение мягких тканей, но кроме этого могут быть и другие дефекты, указывающие на возможное наличие недуга:
- короткий изогнутый внутрь большой палец стопы,
- дефекты в позвоночнике, особенно в шейном отделе,
- короткий большой палец на руках,
- возможно облысение, глухота, замедленное развитие,
- искривление или скос пятого пальца.
Достаточно часто ПОФ провоцирует развитие кривошеи, сколиоза и тугой подвижности костных сочленений.
Если вы заметили у ребенка подобные признаки недуга, то обратитесь к врачу. Доктор, если посчитает нужным, назначит генетическое исследование, в результате которого либо оправдаются подозрения на наличие генной мутации, либо нет.
Диагностика и лечение
Как мы уже упоминали выше, зачастую на ранней стадии фибродисплазии врачи ставят неверный диагноз, что в свою очередь только усугубляет развитие недуга. Обычно болезнь Мюнхеймера диагностируют в возрасте около 14 лет, когда все симптомы становятся явными.
Генетический анализ — единственный способ точно определить наличие изменений в ДНК.
К сожалению, пока болезнь Мюнхеймера считается неизлечимой. Однако американские ученые, проводя исследования, уже выявили, как мы говорили, ген ACVR1, при мутации которого происходит развитие недуга. Сейчас продолжается работа над генными блокаторами, которые должны остановить мутацию. Из-за того, что это заболевание очень редкое довольно сложно проводить исследования.
Несмотря на это, в 2011 году была разработана особая РНК-молекула, которая может убрать поврежденную копию гена ACVR1, никак не влияя на его нормальную копию.
Чтобы проверить, насколько эффективен данный способ, были проведены проверки на стволовых клетках, которые были получены из молочных зубов больных фибродисплазией. В клетках были измененные и нормальные копии ACVR1/ALK2 рецептор-белков. В ходе опыта ученые выяснили, что созданная мРНК способна восстанавливать нормальный синтез белков такой же, как у здоровых людей, нейтрализовав мутировавший ген.
В этом видео рассказывается о том, каким образом проводятся эти анализы и как в принципе выявляются генетические заболевания.
Еще 10 лет назад было невозможно представить, что этот недуг излечим. Сейчас у страдающих болезнью второго скелета появилась надежда на здоровую и счастливую жизнь.
Конечно, предстоит еще большая работа, этот подход к лечению нужно опробовать на лабораторных мышах и только потом перейти к испытаниям на людях. Но, тем не менее, это уже большой шаг. Будем надеяться, что исследования закончатся успешно.
Прогрессирующая оссифицирующая фибродисплазия
Для цитирования: Антелава О.А., Никишина И.П., Гусева И.А., Мякоткин В.А., Хелковская-Сергеева А.Н., Насонов Е.Л., Раденска-Лоповок С.Г. Прогрессирующая оссифицирующая фибродисплазия // РМЖ. 2015. №7. С. 415
Прогрессирующая оссифицирующая фибродисплазия (ПОФ) (от лат. fibro – волокно, dis – расстройство, нарушение, plasis – строение, структура и os, ossis – кость, facio – делать, окостенение) – редкое аутосомно-доминантное заболевание, характеризующееся прогрессирующим гетеротопическим истинным окостенением мягких тканей скелета (соединительнотканных прослоек в толще мышц, фасций, апоневрозов, сухожилий) и ассоциирующееся с множественными врожденными скелетными аномалиями. В МКБ-10 ПОФ представлена отдельной рубрикой – M 61.1.
ПОФ – это редкая нозологическая форма, характеризующаяся рецидивирующими болезненными эпизодами припухлости мягких тканей, ведущими к гетеротопической оссификации через стадию эндохондрального процесса, т. е. превращением соединительной ткани межмышечных прослоек, сухожилий и связочного аппарата в костную ткань [1, 2].
В литературе это заболевание можно встретить под названиями: параоссальная гетеротопическая оссификация, болезнь Мюнхмайера, «болезнь второго скелета», опухоль Стернера [3, 4]. Наиболее часто употреблявшееся ранее название – «оссифицирующий миозит» («превращение мышцы в кость») в настоящее время считается некорректным, поскольку не вполне отражает патогенетические и патоморфологические основы болезни. Общепризнанным научным термином, обозначающий данную патологию, является термин «прогрессирующая оссифицирующая фибродисплазия», что указывает на вовлечение в процесс окостенения соединительнотканных структур вследствие особого характера воспалительных процессов в сухожилиях, связках, фасциях, апоневрозах и мышцах, заканчивающихся окостенением. Расовой, половой и географической предрасположенности к ПОФ не обнаружено [5].
Интерес к этому заболеванию существует с давних времен. Первое упоминание о ПОФ относится к 1648 г., когда Patin описал «окостеневшую» пациентку [6]. В 1996 г. R. Smith et al. проанализировали клиническую картину ПОФ у 28 больных, которых они наблюдали в течение 24 лет. При этом оссификация скелетных мышц начиналась в первые месяцы или годы от рождения с мышц шеи, верхних отделов спины, мышц, окружающих крупные суставы, с последующим распространением на бедра, плечи; поражались также и жевательные мышцы. Примечательно, что скорость прогрессирования болезни и выраженность ограничения движений не зависели от возраста дебюта болезни [7].
Гистопатология пораженных тканей при ПОФ хорошо описана в литературе [8–12]. Исследование тканей ранних проявлений ПОФ говорит об интенсивной мононуклеарной и периваскулярной инфильтрации моноцитов, макрофагов, мастоцитов, T- и B-клеток. Их точная роль в эволюции обострений ПОФ неизвестна, хотя установлено, что любое локальное воспаление независимо от причины его возникновения может спровоцировать активизацию болезни.
Последующая миграция одноядерных воспалительных клеток в пораженную мышечную ткань предшествует масштабной гибели скелетных мышц – процессу, после которого происходит формирование гетеротопического эндохондрального зачатка. За скоротечной воспалительной стадией следует период интенсивной фибробластной пролиферации, сопровождаемой мощным ангиогенезом. Фибробластическое поражение на раннем этапе в отношении гистологии неотличимо от агрессивного ювенильного фиброматоза, но по мере созревания ткани фиброзная ткань проходит через фазу бессосудистого уплотнения до состояния хряща, а затем наступает этап реваскуляризации с остеогенезом и характерным процессом эндохондральной оссификации.
Все стадии гистологического развития активных патологий указывают на то, что различные участки пораженного участка ПОФ проходят полный процесс эндохондральной оссификации с разной скоростью. Примечательно, что образующаяся в результате новая гетеротопическая кость гистологически выглядит как нормальная сформировавшаяся кость и часто содержит включения костного мозга. Мастоциты обнаруживаются на всех этапах формирования тканей при поражении ПОФ, и их количество значительно выше, чем в обычных скелетных мышцах или мышечных тканях больного вне зоны поражения ПОФ. При поражениях ПОФ на этапе фибробластной пролиферации количество мастоцитов многократно превышает показатель, свойственный любой другой форме воспалительной миопатии [13].
При том, что процесс гетеротопного костеобразования в некоторых отношениях схож с формированием костей скелета при эмбриональном развитии или постнатальном заживлении переломов, важнейшим отличием является отсутствие воспалительного компонента при первичном скелетообразовании. Впрочем, возможно, эти различия вполне условны и объясняются недостаточным объемом накопленных данных о морфологии ПОФ на различных стадиях болезни и малой перспективностью их дополнения, поскольку проведение биопсии пораженных (и даже клинически непораженных) тканей, проведение любого, даже малотравматичного хирургического вмешательства сопряжено с исключительно высоким риском образования новых очагов и быстрого прогрессирования болезни. Хотя процесс гетеротропического костеобразования в некоторых отношениях схож с формированием костей скелета при эмбриональном развитии и послеродовом заживлении переломов, важнейшим отличием является отсутствие воспалительного компонента при первичном скелетообразовании.
Генетика ПОФ и семейные исследования. Семейная ПОФ – чрезвычайно редкое заболевание в популяции и имеет аутосомно-доминантный тип наследования с полной пенетрантностью [14]. Хотя большинство зарегистрированных случаев заболевания являются спорадическими (единичными в семье), в литературе описаны семьи, в которых поражены родитель – потомок/потомки: братья, сестры, в т. ч. близнецы. Представляет интерес наблюдение J.M. Connor et al. [15], описавших семью, где ПОФ была диагностирована в 3-х поколениях у 5 лиц. В 1-м поколении пораженным был мужчина, который имел 2-х больных дочерей и 2-х больных внучек. Примечательна выраженная фенотипическая изменчивость тяжести течения заболевания в этой семье. Так, мужчина имел асимптоматическое течение до момента травмы, после которой у него развилась тугоподвижность шеи, спины, конечностей и нижней челюсти. Однако в 70 лет он передвигался, используя трость, и умер в возрасте 72 лет от инфаркта миокарда. Диагноз ПОФ ему был поставлен на основании наличия врожденной деформации большого пальца стопы и рентгенологического обследования, выявившего генерализованный анкилоз позвоночника, множественные зоны оссификации мягких тканей. Одна из его дочерей также не имела признаков заболевания до 22 лет, когда после экстракции зуба с последующим протезированием у нее развилась тугоподвижность челюсти. Ее сестра в возрасте 15 лет отметила спонтанную припухлость левой голени, когда на основании биопсии была диагностирована фибросаркома. Однако дальнейшее течение болезни (характерные болезненные припухлости и прогрессирующее ограничение объема движений) свидетельствовало в пользу ПОФ. Летальный исход наступил в 28 лет от пневмонии в состоянии полной обездвиженности. Также имели ПОФ 2 внучки больного от старшей дочери.
F.S. Kaplan et al. [16] представили данные о передаче ПОФ от пораженного отца 2-м дочерям и сыну. Так, 27-летний мужчина (от брака клинически здоровых родителей, 3-й сын в семье, 2 брата здоровы), имеющий характерную микродактилию большого пальца, заболел в 2 года после травмы. Клинически наблюдалось появление болезненных узлов в области шеи с последующей генерализацией процесса и эктопической оссификацией, прогрессированием болезни и формированием значительного двигательного дефицита. Рентгенологически определялись укорочение и расширение шеек бедренных костей. У 3-х его потомков также присутствовала врожденная микродактилия больших пальцев с дальнейшим развитием типичной картины ПОФ, при этом у одного из его сыновей первые оссификаты четырехглавой мышцы бедра появилась в 2-месячном возрасте после в/м инъекции, у 2-х других детей – в 3-летнем возрасте.
Как свидетельствуют и другие литературные наблюдения [17], авторами часто описывается фенотипическая гетерогенность ПОФ: характер послеродовой гетеротопической оссификации отличается в зависимости от анамнеза, случаев травматизации мягких тканей и заболеваемости вирусными болезнями. По мнению N. Hebela et al. [18], формирование фенотипа заболевания в период пренатального развития определяется генетическими факторами, в то время как сопутствующие (средовые) факторы влияют на послеродовой ход гетеротопической оссификации.
Молекулярная генетика. Е.М. Shore et al. [19] провели полногеномный анализ сцепления на материале 5 семей с ПОФ и выявили сцепление заболевания с регионом 23–24 хромосомы 2 (2q 23-q24), в котором картируется ген ACVR1 (Activine A Rreceptor, type I) (альтернативное название ACK2 – Activine Receptor-like Kinase 2). Секвенирование всех белок-кодирующих экзонов и экзон-интронных областей позволило ученым выявить гетерозиготную мутацию R(206)H (замена аминокислоты аргинин на гистидин в позиции 206) у всех пораженных ПОФ членов 5 семей, а также 32 больных со спорадическим ПОФ. Исследование молекулярно-генетических основ ПОФ в различных популяциях позволило установить, что заболевание тесно сцеплено главным образом с гетерозиготной мутацией de novo R(206)H при спорадической ПОФ [20–22]. Также обнаружена гетерозиготная миссенс-мутация de novo G(356)D в этом же гене [23].
Исследования показали, что во всех случаях спорадических и семейно-наследственных заболеваний у пациентов с классической клинической картиной ПОФ выявлена мутация R(206)H в гене ACVR1/ALK [24–26]. Данный ген кодирует рецептор ACVR1, относящийся к семейству BMP (bone morphogenetic protein)-рецепторов. BMP – регуляторный белок, участвующий в процессе эмбрионального формирования и постнатального восстановления костей скелета (индуцирует развитие мезодермы, формирование костной ткани, зубов, костеобразование и репарацию переломов) [27]. Преобразование сигналов ВМР происходит через рецепторы типа 1: BMPR1А, BMPR1B и ACVR1/ALK2 [28].
A.B. Shafrits et al. [29] обнаружили повышенные уровни белка BMP4 и его экспрессии (мРНК) в лимфобластоидных клетках у 26 из 32 больных с ПОФ и у 1 из 12 здоровых лиц (р A mutation is also recurrent in three Japanese patients with fibrodysplasia ossificans progressiva // J. Hum. Genet. 2007. Vol. 52 (5). P. 473–475.
Последние 2 десятилетия можно рассматривать как переломные в отношении изучения подагры. В.
Прогрессирующая оссифицирующая фибродисплазия (ФОП)
Фибродисплазия относится к редчайшему заболеванию, которое возникает в результате мутации определенного гена. На сегодняшний день в мире зарегистрировано 600 человек, страдающих от него. Патология характеризуется окостенением мышц, связок, сухожилий. Сначала поражается позвоночник, далее страдают другие кости организма.
Характеристика заболевания
Оссифицирующая фибродисплазия (ФОП) по МКБ 10 имеет номер М61.1, возникает из-за генетических мутаций, в результате видоизменения гена ACVR1. Известны случаи, когда поражаются несколько членов семьи, которые находятся в кровном родстве, братья и сестры. Встречается передача заболевания от одного поколения к другому, от отцов к детям.
Данная патология носит название болезни «второго скелета». Прогрессирующую фибродисплазию провоцируют воспалительные процессы мышц, сухожилий, связок, мягких тканей, по истечении времени приводя к окостенению.
Не следует путать данное заболевание с фиброзной дисплазией, при котором происходит замещение здоровой ткани кости на включение костной трабекулы. Фиброзная дисплазия относится к разряду опухолевых состояний, но не является раковой опухолью. Часто происходит перерождение в доброкачественное новообразование, онкология встречается редко.
У пациентов любой ушиб, небольшая ранка не вылечивается, на месте повреждения происходит образование костной ткани.
У новорожденного почти полностью определяется наличие заболевания, от которого одинаково страдают и девочки, и мальчики. Оно обнаруживается загнутым внутрь большим пальцем ноги и отсутствием сустава. Патология проявляется в первые 10 лет жизни больного, характеризуется внезапностью. Нижние конечности страдают в последнюю очередь.
Насколько быстро будет развиваться болезнь установить невозможно. Так как это зависит от количества полученных травм и перенесенных оперативных вмешательств.
Бывают случаи, когда врачи путают оссифицирующую фибродисплазию с онкологией, которая приводит к формированию костных образований. После их удаления происходит резкое усугубление ситуации и ускоренное развитие патологии.
Как развивается
Оссифицирующая фибродисплазия развивается по стадиям:
- На первой стадии образуется небольшой воспалительный очаг;
- На второй стадии патология начинает прогрессировать, воспаление вовлекается в ткани, расположенные около воспаленного места;
- На третьей стадии образуются бессосудистые уплотнения;
- Далее формируется хрящеобразование;
- Завершающей стадией является окостенение.
Оссифицирующая фибродисплазия начинает свое формирование еще в период внутриутробного развития, в результате изменения гена, расположенного во второй хромосоме. Из-за чего происходит мутация формы белка, отвечающего за развитие костной ткани.
Гетеротопические фибродисплазийные очаги обычно формируются в местах повреждения мягкой ткани, ожогов, укусов насекомых, порезов, ушибов. В данной области обнаруживается много лейкоцитов, являющихся фактором иммунного, воспалительного процесса.
Имеются пациенты, которые являются носителями гена, и при определенных обстоятельствах заболевание начинает прогрессировать. На скорость его проникновения оказывает воздействие активность гена. К провоцирующим факторам относятся:
- Хирургические вмешательства;
- Вакцинирование;
- Травмирование кожного покрова;
- Вирусная инфекция
Оссифицирующая фибродисплазия редко начинает развиваться сразу же после рождения ребенка. Врачи обращают внимание на нарушение сформированности пальчиков на ногах и руках. Никаких других внешних проявлений не наблюдается.
Оссификация начинается внезапно, порой без наличия объективной причины. Возраст ребенка может быть от 2-3 месяцев до нескольких лет. Чаще прогрессирование происходит в первые пять лет жизни. Симптомами заболевания считаются:
- Ранняя симптоматика проявляется формированием небольшого уплотнения, размером до 3 мм;
- При прогрессировании уплотнение увеличивается до 10 см;
- При пальпации наблюдается болезненность, иногда покраснение;
- Походка у пациента приобретает неуверенность, скованность;
- На лице отсутствуют какие-либо эмоции;
- Происходит уменьшение объема движений, что имеет связь с появлением новых уплотнений, приводя к полной обездвиженности;
- При прощупывании появляются боли в ногах;
- Нарушается чувствительность;
- Происходят сбои функционирования внутренних органов.
Наиболее очевидной клинической картиной заболевания считают окостенение мягкой ткани. Но возможно проявление следующих дефектов, говорящих о наличии оссифицирующей фибродисплазии:
- Короткий по отношении к остальным большой палец ноги, характеризующийся изогнутостью внутрь;
- Дефект в шейном отделе позвоночника;
- Короткий большой палец рук;
- Отставание в развитии;
- Глухота;
- Облысение;
- Искривление пятого пальца ног;
- Кривошея;
- Сколиоз;
- Тугая подвижность костного сочленения.
Известны случаи, когда уплотнения самостоятельно рассасываются, но в последствии образуются снова. После чего через 2-3 недели происходит их окостенение, в результате которого стает невозможным дальнейшее их рассасывание. У взрослого пациента прогрессирующая болезнь Мюнхеймера приводит к слиянию десятков очагов. Что в последствии вызывает глубокую инвалидизацию и летальный исход.
Диагностика
Диагностирование оссифицирующей фибродисплазии возможно после окончания форсированного костеобразования, которое происходит с момента рождения до 14 лет. Часто заболевание обнаруживается спустя 2 года после его развития. Диагностирование проводится следующими методами:
- Внешним осмотром;
- Рентгенографией;
- Генетической консультацией.
Во время внешнего осмотра у детей до 10-летнего возраста обнаруживаются многочисленные уплотнения, располагающиеся в разных частях тела. В более старшем возрасте определяется пониженная суставная подвижность, искривленный позвоночник, гетеротропические оссификационные очаги. Уплотнения можно увидеть даже невооруженным глазом.
В исключительных случаях может потребоваться гистологическое исследование, но оно проводится редко, чтобы исключить появления новых уплотнений.
Рентгенография полезна на начальном этапе оссифицирующе патологии, поскольку позволяет выявить дисплазию кости, клинодактилию. На поздних стадиях костные уплотнения появляются в мышечной ткани, сухожилиях. Если болезнь зашла далеко, то на рентгеновском снимке не возможно визуализировать внутренние органы.
На сегодняшний день не существует методов, вылечивающих прогрессирующую оссифицирующую фибродисплазию. Но американскими учеными активно проводятся исследования гена, который влияет на развитие патологии. Разрабатываются генные блокаторы, которые способны остановить процесс мутации. Восстанавливается нормальное синтезирование белков за счет нейтрализации мутировавшего гена.
Для лечения оссифицирующей фибродисплазии используют следующие лекарственные препараты:
- Этидронат, который замедляет формирование костной ткани. Но ударная доза средства не только оказывает воздействие на патологические кости, но и приводит к разрушению основного скелета;
- Лекарственные средства, основанные на Ламидроновой кислоте, схожи своим действием с предыдущими препаратами, но не так активно влияют на основные кости;
- Преднизолон оказывает мощное противовоспалительное действие, снижает боль, устраняет опухоли. Курс лечения данным средством не должен превышать 2 недель. Иначе происходит ускорение роста костной ткани, подавление иммунитета, ухудшение зрения;
- Целебрекс замедляет воспаление;
- Метаксалон используется для снижения вспышек заболевания, приводит к ослаблению спазмов.
Улучшает состояние больных, страдающих от фибродисплазии, иммунотерапия Интерфероном. Ее действие направлено на замедление прогресса патологии, уменьшение числа уплотнений. Для облегчения самочувствия пациентам назначают обезболивающие и седативные препараты.
Наибольшую безопасность для больных представляет оперативное вмешательство по поводу устранения уплотнений и костного разрастания. После процедуры обнаруживается усиление процесса окостенения.
На начальном этапе оссифицирующей патологии больному назначается электрофорез на основе йодида калия. Противопоказаны пациентам УВЧ, массажи, парафинотерапия.
Людям с оссифицирующей фибродисплазией необходимо исключение хирургического вмешательства, любых видов инъекций для минимизации риска возникновения новых травм.
Данное заболевание имеет неблагоприятный исход. На продолжительность жизни больных оказывают влияние создание условий, исключающих травм, увечий, инфекционных заболеваний.
Статья написана по материалам сайтов: www.krasotaimedicina.ru, health-post.ru, prosustav.ru, www.rmj.ru, nogivnorme.ru.
»